
Electrodeposition has recently been shown to successfully create three dimensional micro- or nanostructures. Here we demonstrate that highly faceted metallic architectures are readily achieved by appropriate electrodeposition conditions. Three dimensional silver and copper mes-ocrystals with different shapes and size are potentiostatically electrodeposited on highly oriented pyrolytic graphite (HOPG) from simple sulfate and nitrate baths, respectively. The electrodeposition potential plays the most important role in determining the shape and size of mesocrystals. For silver mesocrystals, individual crystals with a range of shapes and sizes are observed which are not influenced by steps in the HOPG substrate. However, when copper is electrodeposited at one distinct potential it tends to form either wires on step edges or particles on terraces. Upon application of a dual pulse potential technique, well defined highly faceted copper mesocrystals are produced.
Introduction
Exploiting the ability of materials to self-organise into well-defined structures is a very active area of study due to the greater flexibility and lower cost of this approach [1–4]. Electrodeposition is one such high-throughput synthesis method which has been widely used to realise structures with different shapes and morphologies. Several electrodeposition techniques have been developed to obtain various morphologies of structures including the template method for nanowires, pulse method for compositionally modulated straures, precursor alloys for granular structures and step-edge decoration for multi-shape structures [5].
Deposit formation at the step edges of some particular substrates such as highly oriented pyrolitic graphite (HOPG) is often termed as electrochemical step-edge decoration [6]. The most important factor determining the growth of metal on a foreign substrate is the energy of the ad-atom and its relationship with the active nucleation sites. The energy of a metallic ad-atom on a carbon or silicon surface is so high that the nucleation of a metallic deposit cannot take place anywhere else but on an active site or along a step edge at the surface. After nucleation, which is mostly instantaneous, further electrodeposition takes place on these grains only, and the deposit remains discontinuous until the nominal (average) coverage achieves a few hundred nanometers [7].
Electrodeposition using this technique has been effectively used for growing structures with a range of sizes, from nano to macro, and to realise regularly shaped crystals of a few metals like Pb, Ag and Pt [8–9]. Indeed, under specific deposition conditions, one can flexibly manipulate the size and the shape of the electrodeposited architectures. However, some metals like Ni have not revealed faceted mesocrytsals due to the incorporation of side reactions such as hydrogen evolution which has a pronounced influence on the deposition mechanism and the produced structures [10–11].
This paper reports the electrochemical deposition of highly faceted mesocrystals of silver and copper on HOPG from simple salt electrolytes and demonstrates the effect of different electrodeposition parameters on morphological evolution.
Experimental
Electrodeposition was carried out in potentiostatic mode in a conventional three-electrode cell connected to a computer-controlled potentiostat (microAutolab III). Commercially available HOPG from SPI Supplies (grade 1) was used as working electrode in a horizontal orientation inside the cell. A platinum plate was used as counter electrode. A Standard Ag/AgCl electrode and high-purity silver wire were used as reference electrodes for electrodeposition of copper and silver, respectively. Electrolytes were prepared by dissolving laboratory grade chemicals into deionized water with a resistivity value of 18 MOhms.cm. The electrolyte was first deaerated with bubbling Ar prior to deposition. To obtain appropriate deposition potentials, cyclic voltammograms were captured in the prepared electrolytes.
The potentiostatic method was used to electrodeposit silver and copper on a HOPG working electrode at ambient temperature. Silver was electrodeposited from an aqueous electrolyte containing silver nitrate with different concentrations at a pH between 2 and 2.5. Copper was electrodeposited from an aqueous electrolyte containing 2 mM copper sulfate. Scanning electron microscopy (SEM, Hitachi 4300SE), Energy dispersive x-ray spectroscopy (EDS, Oxford Instruments) and atomic force microscopy (AFM, Asylum research MFP-3D) were used to study the morphology and structure of the grown materials.
Results and Discussion
1. Electrodeposition of silver mesocrystals
In order to obtain a quick overview of the electrodeposition system AgNO3, cyclic voltammetry was performed on HOPG at different electrolyte concentrations. Figure 1 illustrates typical cyclic voltammograms obtained for silver on a HOPG working electrode which displays the characteristic features of nucleation, namely the large peak separation and the crossover on the cathodic branch. For comparison a cyclic voltammogram for the second sweep taken for each AgNO3 concentration is shown by the dashed line. A negligible difference is observed between the first and second sweeps and any difference is likely reflected by the irreversible nature of the solid state deposition of silver on HOPG. It seems likely that the deposited silver is not readily anodically stripped and therefore promotes the nucleation and deposition of silver in the following cathodic cycle.
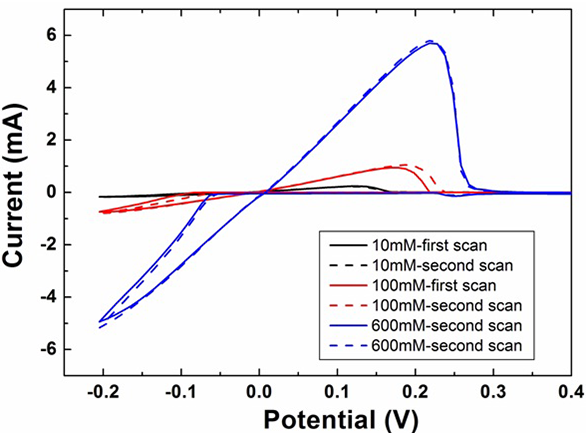
Fig. 1: Typical cyclic voltammograms obtained for Ag on HOPG when depositing from AgNO3 solution with different concentrations
The reduction process occurs mainly in one step revealed by a cathodic reduction peak negative with respect to the formal potential for silver reduction. It has been frequently mentioned [12, 13] that the electrodeposition of silver obeys a diffusion controlled mechanism on carbon substrates. Here we also observe that the reduction peak current increases with increasing the electrolyte concentration and it is reasonable to assume that this corresponds to a mass transport regime.
Chronoamperometric measurements give further details of nucleation and growth of silver on HOPG. Several investigations have discussed this using current transients recorded during the potentiostatic deposition [12–15].
Generally for the current transients for this electrodeposition system, the reaction current rises after a minimum has been reached. The higher the cathodic overpotential applied, the higher is the initial value of the current, the faster its decay, shifting the minima of the current transient curves to shorter times and making the following increase steeper. The rising part of the curves is generally linear and due to the growth of silver nuclei, where charge transfer takes place directly at the growth sites and newly formed silver atoms are immediately incorporated in the crystal lattice. The increase of the current, corresponding to an increase of the reaction surface, lasts until a maximum value is reached. At that time the whole surface is covered with growing silver crystallites [14], and no new active growth sites are formed.
Figures 2 and 3 show the current transients recorded during electrodeposition of silver from 10 mM and 600 mM AgNO3 solutions, respectively, at different deposition potentials. The current transients were stopped before the current starts to drop. This was done since the purpose of our work was to create individual silver centres. The currents corresponding to the rising parts of the current transients are linear in I1/2, indicative of the mass transfer controlled growth of hemispherical nucleation as described by the equation:
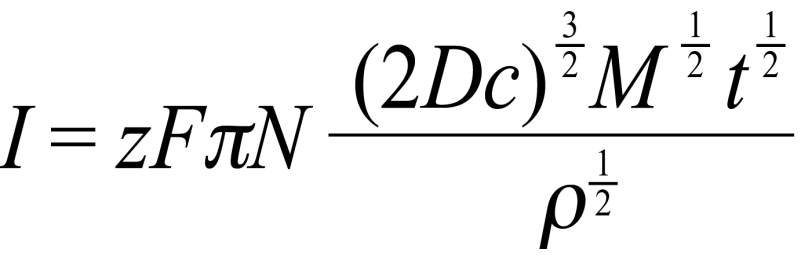
where zF is the molar charge of the electrodepositing species, N is the total number of nuclei, D is the diffusion coefficient, c is the bulk electrolyte concentration, M and r is the molecular weight and density of the electrodeposit, and t is the time. According to the equation, the magnitude of the current, and hence the nucleation rate or N itself, is dependent on various factors. Indeed, in our study we find a strong effect of the deposition potential and also concentration on the current and nucleation rate.
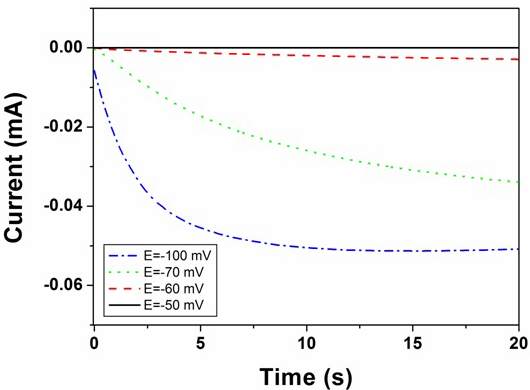
Fig. 2: Current transients recorded during electrodeposition of silver from 10 mM AgNO3 solution at different deposition potentials
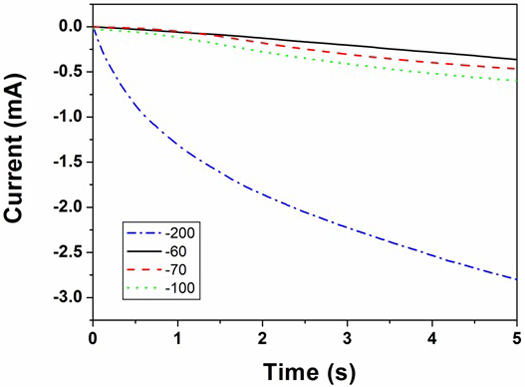
Fig. 3: Current transients recorded during electrodeposition of silver from 600 mM AgNO3 solution at different deposition potentials
Figure 4 shows an SEM image of silver electrodeposited on HOPG at a potential of –0.1 V for 60 s. It is seen that the mechanism of electrodeposition is progressive with the formation of three dimensional silver centres with different sizes and also irregular shapes. It is surprising that surface has not been fully covered by a continuous silver film, instead the nuclei formed grow and become bigger. It has been mentioned [15] that the number of nuclei does not change after a sufficiently long time. A simple explanation of this effect is that the constant number of clusters is determined from the direct exhaustion of active sites on the substrate. Therefore, the distribution and the number of supercritical clusters reflects the number of active sites.
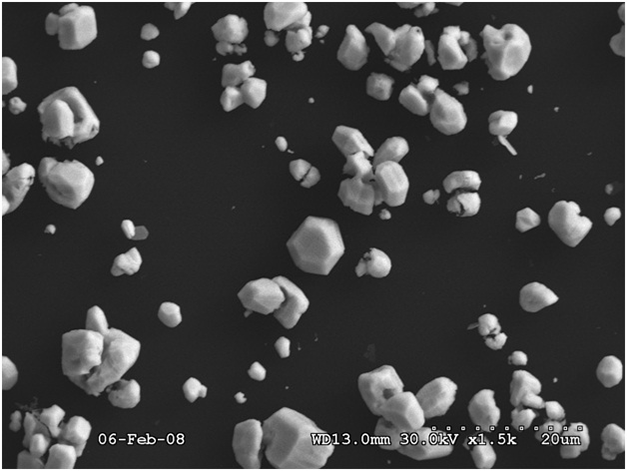
Fig. 4: SEM image of silver electrodeposited on HOPG at a potential of –0.1 V for 60 s reflecting the progressive mechanism of deposition with irregular crystal shapes and sizes
However, this might not be the case, for instance, due to the formation of nucleation exclusion zones arising around growing stable centres. The overlapping of these zones may interrupt the nucleation and thus lead to a saturation nucleus density [15].
On the basis of these findings, we studied the change of morphology of silver electrodeposits by changing the deposition potential, the electrolyte concentration and also the deposition time. Under specific electrodeposition conditions, one can realise highly faceted silver mesocrystals with well defined shapes and size. Figure 5 demonstrates some typical silver mesocrystals realized in our study and achieved under different electrodeposition conditions.

Fig. 5: SEM images of silver mesocrystals electrodeposited at E = –0.1 V from (a) 10 mM AgNO3 for 60 s, (b) 100 mM AgNO3 for 30 s, (c) 250 mM AgNO3 for 10 s and (d) 600 mM AgNO3 for 5 s
2. Electrodeposition of copper mesocrystals
The electrochemical growth of copper from a simple sulphate bath on HOPG was investigated. Figure 6 illustrates a typical cyclic voltammogram taken for copper deposited on a HOPG working electrode from 2 mM CuSO4 solution. It reveals a general behavior of electrodepositing material with a cross over in the cathodic region and separated reduction/dissolution peaks. There is one distinct reduction peak at E = –0.4 V Ag/AgCl and two oxidation peaks at E = +0.29 and +0.47 V Ag/AgCl. The two oxidation peaks seen are assigned to copper stripping (+0.29 V) and oxidation of Cuo to CuO (+0.470 V) [16].
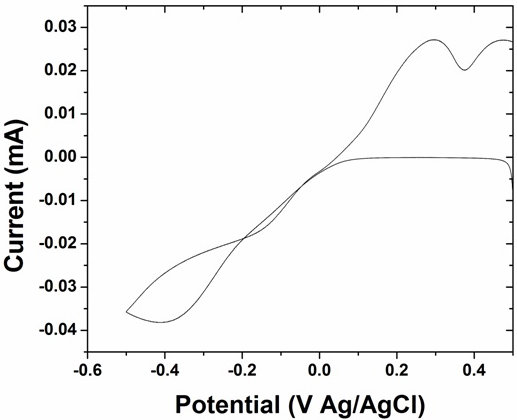
Fig. 6: Typical cyclic voltammograms obtained for Cu on HOPG when deposited from 2 mM CuSO4 solution
We followed the electrodeposition of copper onto HOPG based on the potential-current characteristics achieved during the cyclic voltammetry. Electrochemical step-edge decoration has been investigated in details for several different methods to produce nanoparticles and nanowires. The controllable production of metal electrodeposits has been shown to be possible using a multi-pulse plating method [16]. We employed two different plating pulses including single pulse and dual pulse. Figure 7 shows SEM and AFM images of copper electrodeposited on HOPG at a distinct deposition potential of –0.1 V versus Ag/AgCl. It is seen that copper is electrodeposited in the form of particles and also wires on the HOPG surface. The general form of particles is faceted without great regularity across the whole sample. When the dual pulse technique is employed, three dimensional copper tetragons are formed with same shape and size as shown in Figure 8. The dual pulse technique permits the electroactive species to add to the metallic centre and grow in a regular form. The first short pulse corresponds to the nucleation stage and the second long pulse corresponds to the growth of three dimensional copper mesocrystals.

Fig. 7: SEM and AFM images of copper electrodeposited from 2 mM on HOPG at a distinct deposition potential of –0.1 V Ag/AgCl for 60 s

Fig. 8: SEM and AFM images of copper pulsed electrodeposited from 2 mM on HOPG at –0.1 V Ag/AgCl for 5 ms and –0.055 Ag/AgCl for 600 s
Conclusion
Truly three dimensional silver and copper mesocrystals were realized by electrochemical deposition. Simple salt aqueous solutions were used as electrolytes to create metal centres using single and dual pulse electrodeposition on HOPG. Silver is electrodeposited on HOPG as individual non-overlapping islands. The shapes and size of silver mesocrystals are highly influenced by the potential applied and the electrodeposition technique.
- Ahmadi, T. S.; Wang, Z. L.; Green, T. C.; Henglein, A.; El-Sayed, M. A. Science 1996, 272, 1924–1926.
- Xu, Z.; Xiao, F. S.; Purnell, S. K.; Alexeev, O.; Kawi, S.; Deutsch, S. E.; Gates, B. C. Nature 1994, 372, 346–348.
- Bruchez, M., Jr.; Moronne, M.; Gin, P.; Weiss, S.; Alivisatos, A. P. Science 1998, 281, 2013–2016.
- Bezryadin, A.; Lau, C. N.; Tinkham, M. Nature 2000, 404, 971–974.
- Péter L. und Bakonyi I., Electrodeposition as a Fabrication Method of Magnetic Nanostructures in: Nanomagnetism and Spintronics, Kapitel 5, Hrsg. F. Narirpouri und A. Nogaret, World Scientific Publisher, 2010.
- Penner RM, J. Phys. Chem. B, 2002, 106, 3339-3353.
- M. Ohring, Materials Science of Thin Films, Academic Press, USA, 2000.
- Zach MP, Ng K, Penner RM, Science, 290 (2000) 2120–2123.
- Xiao ZL, Han Y, Kwok WK, Wang HH, Wlp U, Carbetree GW, J. Am. Chem. Soc., 2004, 126, 2316–2317.
- Nasirpouri F, Bending S, Peter L, Fangohr H, , Thin Solid Films, 519 (2011) 8320–8325.
- Nasirpouri F, Engbarth M, Bending S, Peter L, Knittel A, Fangohr H, Milosevic M, Applied Physics Letters, 2011, 98, 222506.
- Gunawardena G, Hills G, Montenegro I, J Electroanalytical Chemistry, 1982, 138, 241–254
- Schmidt U, Donten M, Osteryoung J, J Electrochem Soc, 1997, 144, 2013–2021
- Simons W, Gonnissen D, Habin A, J Electroanalytical Chemistry, 1997, 433, 141–151.
- Milchev A, Electrochimica Acta, 1983, 28, 947–953
- Walter EC, Murray BJ, Favier F, Kaltenpoth G, Grunze M, Penner RM, J. Phys. Chem. B, 2002, 106, 11407–11411.
PDF Version of the article |
Flash Version of the article |
|
| [qr-code size=”2″] | ||